
La protéine p53 est l’effecteur d’un point de contrôle essentiel au maintien de l’intégrité du génome et au contrôle de la division cellulaire, de la sénescence et de la mort cellulaire, qui est fréquemment mutée au cours du développement tumoral. Ses fonctions suppresseur de tumeur ont plus récemment été étendues à la régulation du métabolisme. Plusieurs laboratoires, dont le nôtre, ont montré que différents régulateurs clés de la voie impliquant la protéine p53 (MDM2, MDM4, E4F1, ARF, BMI1) contrôlent également le métabolisme cellulaire de façon dépendante et indépendante de p53. L’objectif de notre laboratoire est de mieux comprendre les réseaux métaboliques régulés par la voie p53 et comprendre comment leur perturbation contribue à différents processus physiopathologiques incluant certaines maladies métaboliques, le vieillissement et le cancer.
Axe 1 : Modélisation des réseaux métaboliques régulés par la voie p53 (Project Leader : L. Le Cam)
Nous avons entrepris de cartographier les réseaux métaboliques régulés par la voie p53 à un niveau sans précédent en caractérisant par une stratégie multi-omics (RNAseq, proteomics, metabolomics) des cellules primaires dérivées d’une collection unique de modèles murins génétiquement modifiés dans lesquels un, deux ou trois composantes de la voie p53 (Mdm2, Mdm4, E4f1, Trp53) sont génétiquement inactivés ou qui expriment différents mutants de p53 fréquemment retrouvés dans les tumeurs humaines (R175H et R248W). Ces larges sets de données sont intégrés dans des modèles computationnels que nous utilisons pour identifier de nouveaux biomarqueurs ou définir de nouvelles stratégies thérapeutiques
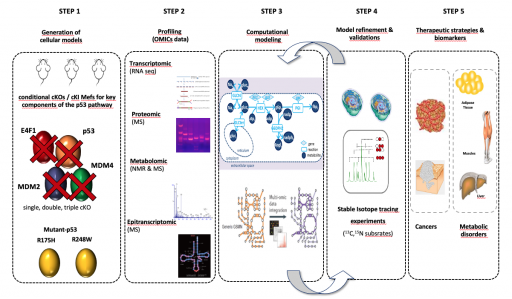
Axe 2 : Rôle des formes mutées de p53 dans le syndrome Li-Fraumeni (Project Leader : L. Le Cam)
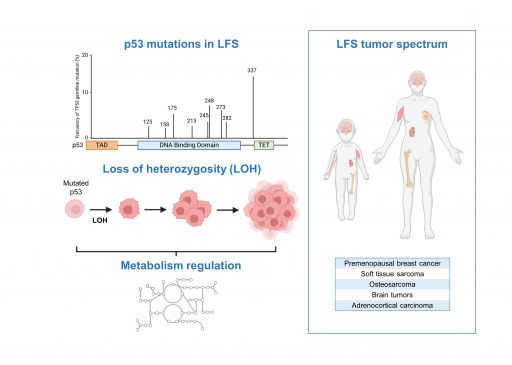
Les mutations germinales du gène TP53 sont associées à un syndrome de prédisposition tumoral sévère appelé le syndrome Li-Fraumeni (LFS). Au travers de la caractérisation d’échantillons de patients LFS et de nouveaux modèles animaux récapitulant certaines des altérations génétiques communément retrouvées dans le LFS, notre objectif est de mieux comprendre comment certaines formes mutées de la protéine p53 régulent le métabolisme au cours de la progression tumorale.
Axe 3: Dérégulation des fonctions métaboliques de p53 au cours du vieillissement (Project leader : PF. Roux)
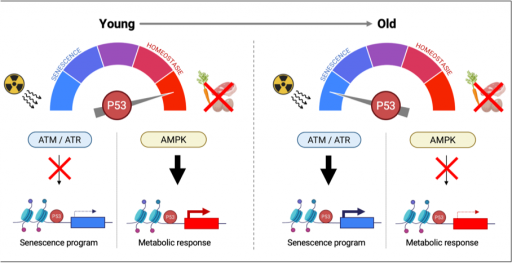
La voie p53 joue un rôle central dans la régulation des mécanismes impliqués dans le vieillissement, tant au niveau cellulaire que de l’organisme entier. Elle intervient également dans l'adaptation métabolique face à des challenges nutritionnels physiologiques comme ceux intervenant au cours du jeûne. À l'aide d'approches de biologie des systèmes, nous explorons comment le vieillissement influence la capacité de la voie p53 à réguler le métabolisme, contribuant ainsi à l’émergence de certaines pathologies liées à l'âge tel que le diabète de type II.
Axe 4: Rôle de l’hétérogénéité métabolique dans les tumeurs cutanées (Project leaders : M. Lacroix, E. Frouin, PE Stoebner).
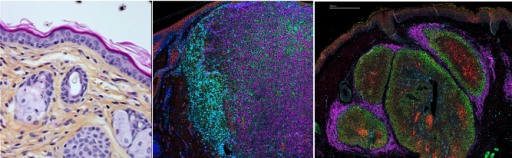
Dans cette partie du projet d’équipe, nous adressons le rôle de différents réseaux métaboliques régulés par les acteurs de la voie p53 dans l'homéostasie cutanée et comment leur perturbation contribue à la reprogrammation des cellules cancéreuses de différents types de cancers de la peau (mélanomes, carcinomes sébacés). Nous utilisons des échantillons de patients et de différents modèles murins (PDXs et modèles génétiquement modifiés) que nous caractérisons à travers des approches d'imagerie multiplexées in situ (Hyperion, Xenium) pour évaluer l’hétérogénéité tumorale sous l’angle du métabolisme. Un autre de nos objectifs est d’évaluer si la zonation métabolique intra-tumorale définit des niches spécifiques de certaines sous-populations tumorales qui contribuent à la diffusion métastatique et à la résistance aux traitements.
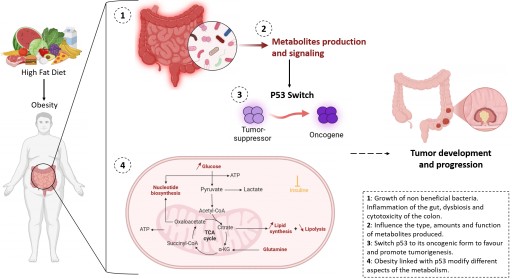
Axe 5: Echanges métaboliques entre tumeurs et l’hôte (Project leader : M. Lapierre)
Au cours de l'initiation et de la progression tumorale, de nombreux changements métaboliques influencent la fonction des cellules tumorales qui leur permettent de survivre dans des conditions défavorables (hypoxie, privation de nutriments...). Des échanges de métabolites interviennent entre les cellules du microenvironnement tumoral mais aussi entre cellules cancéreuses et d’autres organes à plus longue distance. Cet axe aborde la façon dont certains changements métaboliques intervenant dans le foie et/ou le tissu adipeux, notamment ceux liés à l’obésité, façonnent à distance l'écosystème de tumeurs colorectales mutées pour le gène p53 en utilisant une combinaison de modèles animaux originaux, de tumoroïdes et d’échantillons de patients. Ce projet fait également appel à différentes méthodes d'imagerie multiplexées que l’on utilise pour évaluer le métabolisme in situ.
Axe 6: Reprogrammation métabolique des tumeurs IDH mutées (Project leader : L. Stuani)
Dans cet axe, nous visons à élucider les mécanismes sous-jacents aux spécificités métaboliques, ainsi que leurs conséquences moléculaires, des cancers porteurs de mutations de l'Isocitrate Déshydrogénase (IDHm), notamment les gliomes (oligodendrogliomes et astrocytomes) et les Leucémies Aiguës Myéloïdes (LAM). Par ailleurs, il a été récemment rapporté que le spectre des mutations de TP53 dans les cancers IDHm est dominé par une mutation « hotspot » conduisant à l’expression d’une forme mutée p53R273C. A l’aide de modèles expérimentaux, nous essayons de comprendre le rationnel de cette pression de sélection conduisant à cette combinaison d’évènements génétiques. Nos objectifs sont également d’identifier de nouvelles approches thérapeutiques adaptées à ce sous-groupe de patients, et de mieux comprendre l’hétérogénéité et la plasticité métaboliques de ces cellules cancéreuses dans différents écosystèmes.


Situé dans le Parc Euromédecine, l'Institut de Recherche en Cancérologie de Montpellier (U1194) est localisé sur le Campus Val d'Aurelle (ICM, Institut régional du Cancer Montpellier / Val d'Aurelle).
Centre de Recherche U1194 soutenu par l'Inserm, l'Université de Montpellier et l'Institut du Cancer de Montpellier (ICM)
 Institut de Recherche en Cancérologie de
Institut de Recherche en Cancérologie de


